-
 闵行区仿制药eCTD推荐04.11
闵行区仿制药eCTD推荐04.11此次eCTD实施范围的扩大对外企而言影响。实施范围的扩大为外企提供了更多选择,特别是在产品线中NDA和AND占比相当的情况下。外企的系统和流程相对成熟,因此它们对eCTD扩大范围持积极态度,更愿意尝试...

-
 浦东新区生物制品eCTD找哪家04.10
浦东新区生物制品eCTD找哪家04.10美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...
-
 福建瑞士eCTD04.10
福建瑞士eCTD04.10美国电子提交通道ESG(Electronic Submissions Gateway)是美国食品药品监督管理局(FDA)建立的电子化监管信息提交系统,旨在为制药、生物制品、医疗器械等行业提供安全、高效...
-
江苏生物制品eCTD04.10
eCTD的实施为监管机构和企业带来了多重机遇。电子化申报资料能够极大地加速审评效率,减少人为判断错误和数据混淆的情况,从而提高审评的准确性和速度。同时,eCTD带来的数据标准化机遇使得全球监管机构的资...
-
 徐汇区国产eCTD发布软件04.09
徐汇区国产eCTD发布软件04.09美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...
-
 国产eCTD业务04.09
国产eCTD业务04.09生命周期管理与变更递交 eCTD支持全生命周期管理,申请人需通过序列更(Sequence)反映药品变更信息。例如CEP证书的更需提交“变更说明表”,对比已批准和拟修改内容,并附修订版技术文档。重大变更...
-
 浙江INDeCTD是什么04.09
浙江INDeCTD是什么04.09美国药物主文件(Drug Master File, DMF)是向FDA提交的机密技术文件,用于支持药品生产、质量控制及合规性审查。以下为申报的要点和流程总结: DMF概述与类型 定义与作用 DMF是...
-
 重庆eCTD系统04.08
重庆eCTD系统04.08美国于2003年成为全球早采用eCTD(电子通用技术文档)的国家之一,初由CDER和CBER作为电子提交平台试点。2008年起,eCTD正式成为药申请(NDA)和生物制品许可申请(BLA)的标准格式,...
-
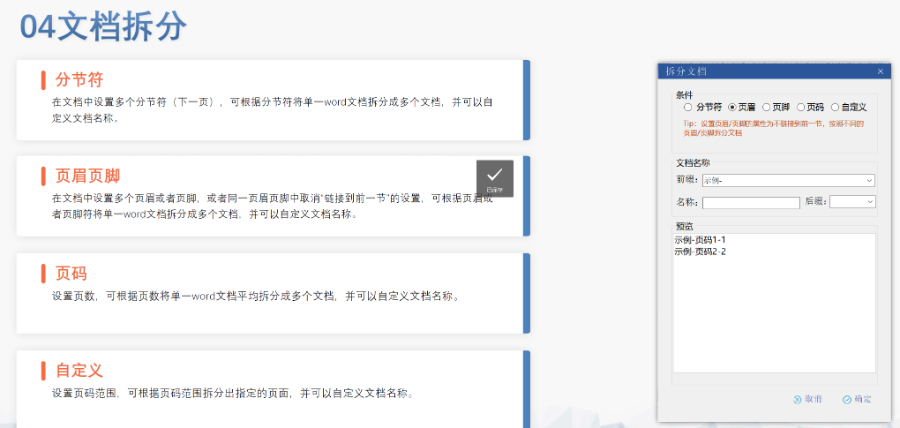 徐汇区仿制药eCTD服务放心可靠04.08
徐汇区仿制药eCTD服务放心可靠04.08申报流程与要求 资料准备 内容要求:包括产品描述、生产工艺(原材料来源、设备参数等)、质量控制标准(SOP、稳定性数据)、安全性与毒性研究等。 格式规范: 采用CTD(通用技术文件)格式,按模块...
-
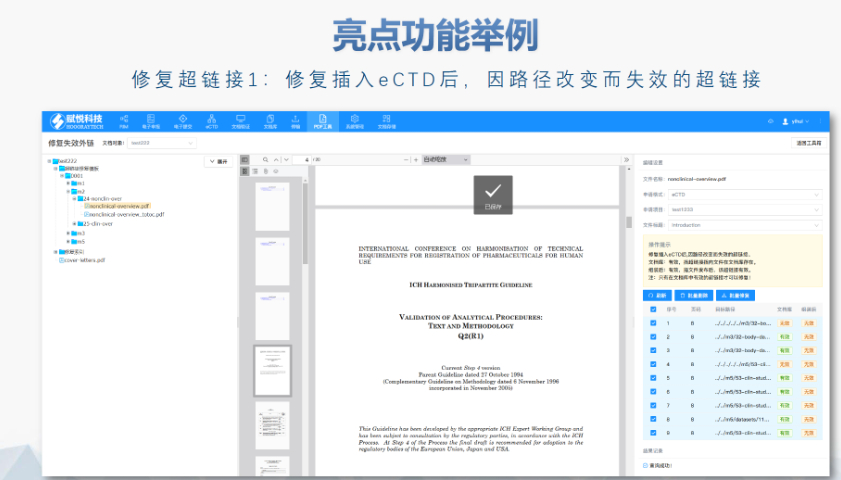 湖北INDeCTD04.07
湖北INDeCTD04.07ANDA递交: 按照ICH M4的CTD格式整理资料,并以eCTD格式递交; 通过ESG通道递交资料; 收到CDER的letter,说明资料已经进入FDA数据库; 付GDUFA费,在资料递交后的10日...
-
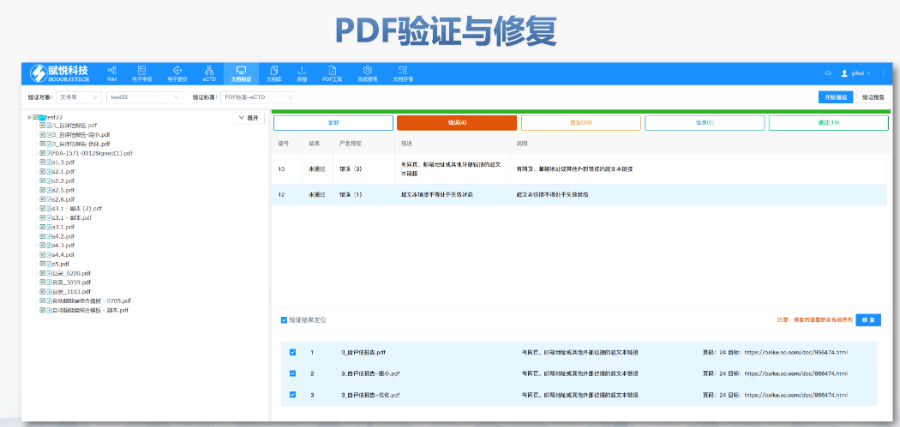 工业园区生物制品eCTD文件如何制作04.07
工业园区生物制品eCTD文件如何制作04.07仿制药作为提高药物可及性与可负担性的一类药物,2012年以前,注册审评是不收取任何费用的,但当时仿制药申请积压严重,从申报到获批需要3~5年的时间。 美国国会于2012年颁布了仿制药使用者费用修正案(...
-
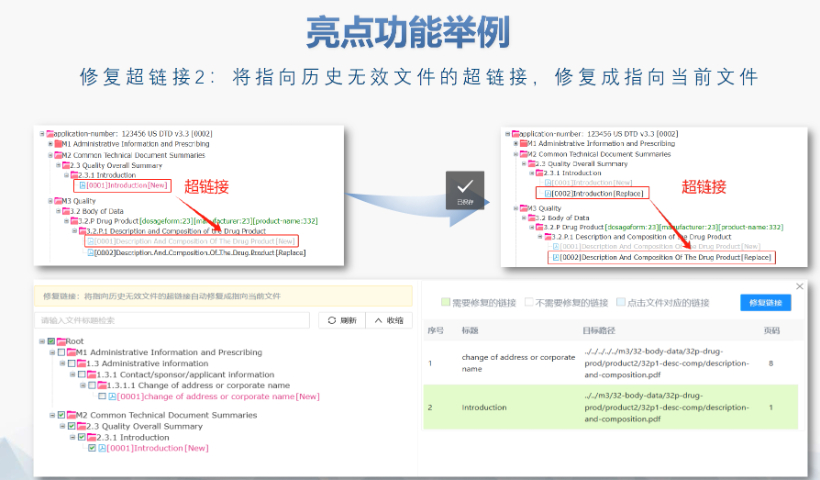 黑龙江eCTD品牌04.07
黑龙江eCTD品牌04.07欧洲药品管理局:集中审评程序由欧洲药品管理局(European Medicines Agency, EMA)负责协调。 人用药品委员会:人用药品委员会(Committee for Medicinal ...