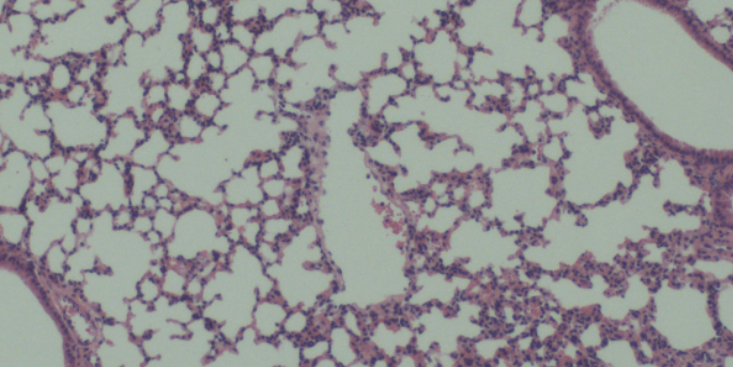
原理过表达稳定细胞系(OverexpressionStableCellLines)的构建利用慢病毒转染、电转等技术手段将特定的基因序列整合到宿主细胞的染色体上,使得目的基因能够在宿主细胞内长期且稳定的表达。用途基因功能研究、免疫/杀伤/增殖等、抗体药物的活性筛选(细胞水平的结合和阻断)、CAR分子的杀伤活性评价。材料与仪器(以慢病毒pMSCV载体为例)包含目的基因和eGFP-Tag的pMSCV质粒、带有eGFP-Tag的pMSCV空载质粒、GAG质粒、VSV质粒、Puromycin、Polybrene、Lipo3000、HEK293T细胞、opti-MEM、DMEM、FBS、双抗、μm的滤膜、荧光显微镜等。步骤1、基因的构建1)根据目的基因mRNA编码区设计引物,分别在引物两端加入酶切位点EcoRI和BglII。2)从细胞中提取目的基因mRNA,然后逆转录成cDNA,然后从cDNA里面用引物把目的基因的CDS区扩增出来。PCR扩增出带有酶切位点的目的基因编码区序列,连接至pMD19-T载体后转化至感受态细菌DH5α,分别进行菌落PCR鉴定和酶切鉴定。3)使用EcoRI和BglII双酶切下目的基因序列,电泳割胶回收纯化,连接到pMSCV-eGFP载体。再次转化到感受态细菌DH5α,菌落PCR鉴定和酶切鉴定成功后,送至公司测序、鉴定。4)鉴定成功后,将质粒转化至感受态细菌DH5α中。因此,对动脉血管平滑肌细胞的体外培养和研究可用来发现和确定新的血管疾病的靶向**方法。宁夏成瘤原代细胞分离培养说明书
细胞增殖通俗点讲,细胞增殖也就是细胞分裂,就像我们每个人的生命起点都是由一个受精卵不断的分裂而来,然后形成由非常多的细胞组成的个体,当然在增殖期间也不断有衰老和死亡的细胞出现,这就是细胞增殖。增殖是生物体生长、发育、繁殖及遗传的基础。大家了解细胞增殖说明了什么吗?细胞增殖的状态是什么样的?上海东寰给大家讲解一下。细胞增殖简单来说,只要有增殖就说明细胞还活着,所以细胞增殖是生物体的重要生命特征。科研小伙伴研究它干什么呢?举几个例子,比如某小伙伴发现了一种可能杀死肿的小分子,那如果要验证这个小分子是否有效,那就要研究加入这个小分子后的肿细胞增殖情况,又比如某个科研小伙伴突发奇想,把细胞的某段基因给切了,想看看对这个细胞有什么影响,是没变化还是活的更久,亦或者细胞死的更快等等,这些研究都要来检测细胞的增殖。怎么知道细胞的增殖状态呢?随着生物科技不断地发展,出现了很多检测方法,简单来说一种是直接法,这种方法就是直接测定进行分裂的细胞数来评价细胞的增殖能力,还有一种是间接的方法,我们通过检测细胞的活力来评价细胞的增殖能力,比如我们常见的染色法MTT、CCK8、流式法等等,也有通过不染色不标记的设备。湖南原代细胞分离培养科研用的原代细胞哪里有卖的。
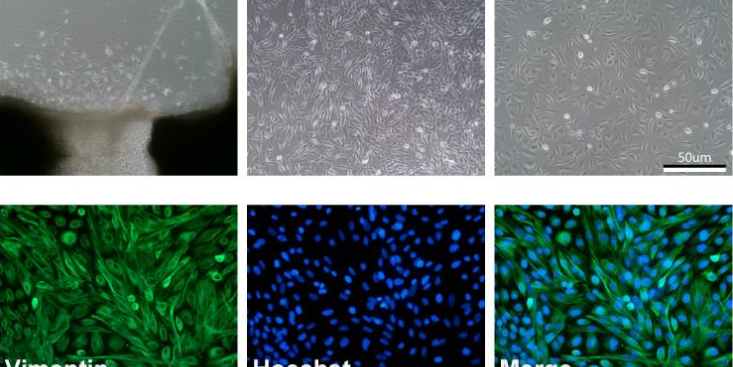
并进质粒抽提。GAG质粒和VSV质粒同样可以转化至感受态细菌DH5α中,并进行无内质粒抽提。质粒抽提后冷冻于-20℃保存。2、慢病毒包装1)使用DMEM完全培养基培养6cm皿HEK293T至汇合度为70~80%。采用opti-MEM和Lipo3000分别转染含有目的基因的pMSCV-eGFP、VSV、GAG质粒及对照载体,每皿加入脂质体-质粒转染混悬液按购买脂质体相关说明书操作定量。继续培养24h。2)24小时后,将培养基更换为新鲜的DMEM完全培养基,放进细胞培养箱继续培养48~72h。3)48~72h后收集上层培养液,并过μm滤膜,采用ELISA法对所获得的慢病毒载体进行病毒滴度测定。如不及时使用可以冻存于-80℃。3、慢病毒转染1)转染前1天将细胞接种6孔培养板,时细胞的融合率约为50%,前需换液,加入1mLDMEM完全培养基。2)病毒冰浴融化后加入相应体积的病毒液及聚凝胺(Polybrene),混匀后放入37℃孵箱中继续培养3)4h后补充1mL培养基,14h后换液(24h内换液即可)。4)病毒72h后用倒置显微镜观察荧光,监测效率,出现较多荧光时将等量的转染细胞和未转染细胞分别加入等浓度Puromycin(Puromycin或其他筛选浓度需要事先摸索)。5)待未转染细胞全部死亡并且可观察到满意荧光量时,降低Puromycin浓度培养。
一、原理在体外培养皿或平板培养的单层贴壁细胞上,用微量头或其它硬物在细胞生长的区域划线,去除部分的细胞,然后继续培养细胞至实验设定的时间,取出细胞培养板,在显微镜下观察并拍照记录特定位置细胞的生长迁移能力。通过不同分组之间的细胞对于划痕区修复能力的不同,可以判断各组细胞的迁移与修复能力的差别。二、步骤1、准备:所有能灭菌的器械都要灭菌,直尺和marker笔在操作前紫外照射30min(超净台内)2、流程:1.培养板划线:先用marker笔在6孔板背后,用直尺比着,均匀得划横线,大约每隔cm一道,横穿过孔,每孔至少穿过5条线;2.铺细胞:在孔中加入约5×105个细胞,具体数量因细胞不同而不同,掌握为过夜能铺满;3.细胞划线:第二天用头比着直尺,尽量垂至于背后的横线划痕,头要垂直,不能倾斜;4.洗细胞:用PBS洗细胞3次,去除划下的细胞,加入无血清培养基;5.细胞培养及观察:放入37℃、5%CO2培养箱,培养;按0,6,12,24小时取样,倒置显微镜观察特定位置细胞迁移情况并拍照;6.结果分析:使用ImageJ软件打开图片后,随机划取6-8条水平线,计算细胞间距离的均值。三、注意事项1、铺细胞使用6孔板,因为6孔板可以保证有相当距离的平直划痕。小鼠的肝细胞通常是指小鼠的肝实质细胞。

使其形成利于核酸进入的微孔。C.逆转录病毒(RNA):通过病毒中膜糖蛋白和宿主细胞表面的受体相互作用进入宿主细胞,之后反转录酶启动合成DNA并随机整合到宿主基因组中。特点:稳定转染,可用于难转染的细胞、原代细胞,体内细胞等,但携带基因不能太大。D.腺病毒(双链DNA):先和细胞表面的受体结合,继而在αV整合素介导下被细胞内吞。特点:可用于难转染的细胞。2、影响转染的因素血清:血清影响复合物的形成,降低转染效率。阳离子脂质体和DNA的量在使用血清时会有所不同,因此想在转染培养基中加入血清时要进行条件优化。大部分细胞可以在无血清培养基中几个小时内保持健康。:比如青霉素和链霉素,是影响转染的培养基添加物。这些一般对于真核细胞无毒,但阳离子脂质体试剂增加了细胞的通透性,使可以进入细胞。这降低了细胞的活性,导致转染效率低。细胞代数:转染前细胞经过1-2次传代保证细胞生长旺盛容易转染,注意贴壁细胞一旦长满就不好转染。细胞铺板密度:一般转染时,贴壁细胞密度为70%-90%,悬浮细胞密度为2*10^6--4*10^6细胞/ml时效果较好。确保转染时细胞没有长满或处于静止期。铺板细胞数目的增加可以增加转染活性和细胞产量。肝脏星状细胞占肝脏固有细胞总数的15%,占非实质细胞的30%左右。安徽怎样原代细胞分离培养厂家
足细胞呈星型多突状,胞体较大,由胞体伸出许多突起,呈指状交叉覆盖于肾小球基底膜外表面。宁夏成瘤原代细胞分离培养说明书
如何判断是否加入了合适体积的Matrigel:我们只需用一张格子纸就能知道自己的移液调整到多少能正好把下孔填满。如上图所示,垂直透过每个孔看下面的格子纸,如果格子被缩小了,那么就说明胶没加满,格子被放大了,那么胶就加多了,格子没发生什么变形,这才是刚刚加满下孔的状态。2、凝胶1)盖上ibidi血管生成载玻片的盖子。2)准备一个10cm的培养皿,放入浸过水的纸巾,制成一个湿盒。3)将ibidi血管生成载玻片放入培养皿中,盖上培养皿盖。4)将整个培养皿放入培养箱中,静置30分钟左右,等待胶凝结。5)等待同时准备细胞悬液。3.铺细胞加入细胞的量直接影响实验结果,所以在正式实验开始之前,要对不同类型的细胞和使用数量进行预实验。获得比例的细胞密度。我们的实验使用HUVEC细胞,每孔种10000个细胞即可。1)准备密度为2×105的细胞悬液,充分混匀。2)将胶已经凝固的ibidi血管生成载玻片从湿盒中取出。3)每孔加入50μl的细胞悬液,注意保持头垂直在上孔的上方,不要接触下孔的凝胶。可以使用排。4)同样用格子纸查看是不是加了足够量的液体,如果没有,加入无细胞的培养基,使上孔液体正好加满。5)盖上盖,静置,一段时间后。宁夏成瘤原代细胞分离培养说明书
使每条染色体的着丝点排列在细胞中间的一个平面上。这个平面与纺锤体的中轴相垂直,类似于地球上赤道的位置...
【详情】细胞增殖通俗点讲,细胞增殖也就是细胞分裂,就像我们每个人的生命起点都是由一个受精卵不断的分裂而来,然...
【详情】日久天长,易发生如下变化:分化现象减弱;形态功能趋于单一化或生存一定时间后衰退死亡;或发生转化获得不...
【详情】原理以慢病毒包装为例,主要包括3个质粒:质粒DNA可以转录出慢病毒遗传物质(RNA),但不能翻译出慢...
【详情】流式细胞检测是一种应用于生物医学研究和临床诊断的技术。接下来就由上海东寰为您介绍。它通过将细胞悬浮液...
【详情】细胞迁移和侵袭实验细胞迁移和侵袭技术服务(DH0004)一、服务介绍细胞迁移\侵袭是指细胞在接收到信...
【详情】病毒包装技术服务病毒包装技术服务(DH0010)一、服务介绍重组病毒以其高效和多样性,是非常有效的将...
【详情】日久天长,易发生如下变化:分化现象减弱;形态功能趋于单一化或生存一定时间后衰退死亡;或发生转化获得不...
【详情】使其形成利于核酸进入的微孔。C.逆转录病毒(RNA):通过病毒中膜糖蛋白和宿主细胞表面的受体相互作用...
【详情】食品中的大肠杆菌进行快速准确的检测已成为了人们经常关注的问题。下面阐述食品中的大肠杆菌检测的方法及分...
【详情】使每条染色体的着丝点排列在细胞中间的一个平面上。这个平面与纺锤体的中轴相垂直,类似于地球上赤道的位置...
【详情】含血清完全培养液在2-8℃保存,需在1周内用完;5.增加接种细胞起始浓度;6.换用新的保种细胞;7....
【详情】